Diagnostik
Die Diagnose einer CPAM, also einer kongenitalen zystadenomatoiden Malformation der Lunge, erfolgt üblicherweise noch bevor das Kind geboren wird, mithilfe bildgebender Verfahren während der Schwangerschaft. Hier sind die Schritte, die üblicherweise unternommen werden, um eine CPAM zu diagnostizieren:
1.
Ultraschalluntersuchung: Dies ist die häufigste Methode zur Erkennung einer CPAM. Während der routinemäßigen pränatalen Ultraschalluntersuchungen können wir auffällige zystische Formationen oder abnorme Massen in den Lungen des Fötus feststellen. Der Ultraschall bietet gute Bilder, um die Größe und die Lage der CPAM zu beurteilen.
2. Magnetresonanztomografie (MRT): Wenn die Ultraschallbilder nicht eindeutig sind oder eine detailliertere Darstellung erforderlich ist, kann in unserer Klinik eine fetale MRT durchgeführt werden. Diese Technik bietet präzisere Bilder der fetalen Lunge und kann helfen, CPAM von anderen möglichen Lungenanomalien zu unterscheiden.
3.
Nachgeburtliche Untersuchungen: Nach der Geburt können weitere Tests durchgeführt werden, um die Diagnose zu bestätigen und den Zustand detailliert zu bewerten. Dazu gehören:
·
Röntgenaufnahme der Brust: Dies kann helfen, die Größe und den Zustand der CPAM nach der Geburt zu beurteilen.
- Magnetresonanztomografie (MRT) der Brust: Ein MRT-Scan bietet detaillierte Bilder der Lunge und kann genutzt werden, um die Struktur der CPAM und ihre Beziehung zu anderen Lungenbereichen zu verstehen. Sie benötigt keine für das Kind belastende Röntgenstrahlung
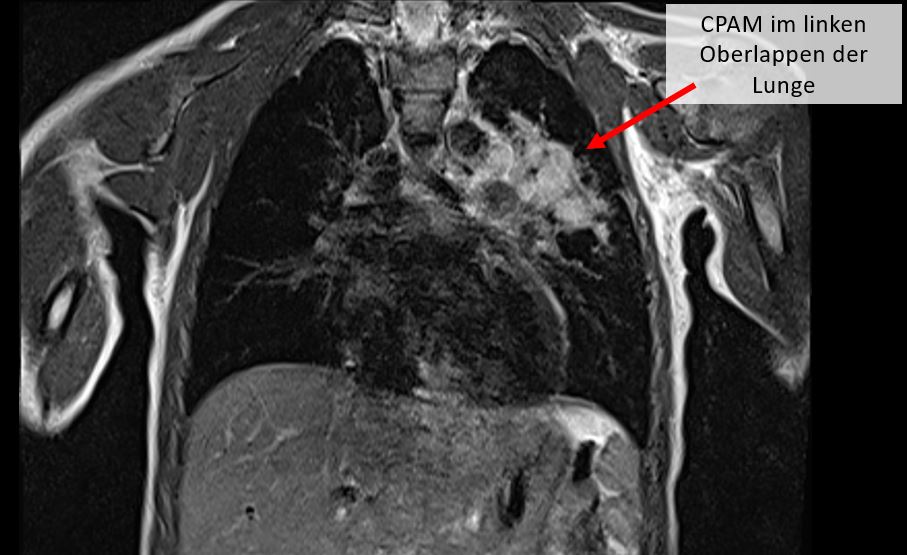
- Echtzeit-Magnetresonanztomografie („real-time-MRT) der Brust: Die Real-Time-MRT (Echtzeit-Magnetresonanztomografie) ermöglicht es, dynamische Vorgänge im Körper sofort zu visualisieren, wie beispielsweise das schlagende Herz oder Atembewegungen. Diese Technik ist besonders wertvoll für die Überwachung von Organen während der Bewegung und kann auch während chirurgischer Eingriffe genutzt werden, um uns eine präzise Führung zu bieten. Sie kann sogar beim Neugeborenen angewendet werden.
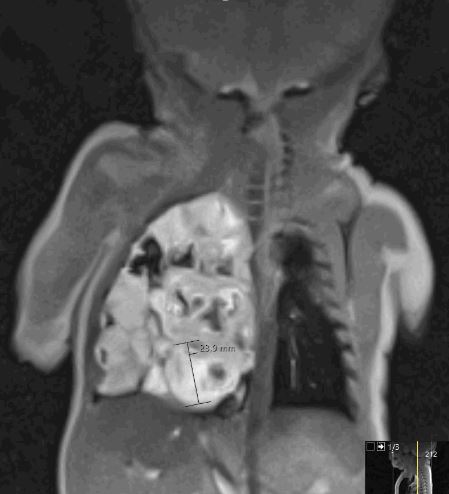
·
Computertomografie (CT) der Brust: Ein CT-Scan bietet ebenso detaillierte Bilder der Lunge und kann genutzt werden, um die Struktur der CPAM und ihre Beziehung zu anderen Lungenbereichen zu verstehen. Die damit verbundene Röntgenstrahlung ist jedoch nicht unerheblich, daher führen wir eine CT nur in Ausnahmefällen durch.
4. Beobachtung und Bewertung: Zusammen mit den Neonatologen überwachen wir auch die Atmung und das allgemeine Wohlbefinden des Neugeborenen, um festzustellen, ob die CPAM Symptome verursacht, die eine sofortige Behandlung erfordern.
Diese Diagnosewerkzeuge ermöglichen es uns, eine genaue Diagnose zu stellen und den besten Behandlungsplan für das Kind zu entwickeln, abhängig von der Schwere und den Symptomen der CPAM.
Therapie
Die Behandlung einer CPAM (Congenital Pulmonary Airway Malformation) hängt von verschiedenen Faktoren ab, darunter die Größe und Lage der Malformation sowie das Vorhandensein von Symptomen. Hier sind die gängigen Ansätze zur Behandlung von CPAM:
1.
Überwachung: Bei asymptomatischen Fällen, insbesondere wenn die CPAM klein ist und keine Atemprobleme verursacht, kann auch eine abwartende Haltung eingenommen werden. Regelmäßige Nachuntersuchungen mit Röntgen, ggf. Schnittbildgebung (MRT oder Ultraschall) sind erforderlich, um sicherzustellen, dass sich die CPAM nicht vergrößert oder Komplikationen verursacht.
Das Überwachen einer CPAM ohne chirurgische Eingriffe kann sicher sein, wenn die CPAM klein, nicht wachsend und symptomfrei ist. Entscheidend sind regelmäßige medizinische Untersuchungen zur Kontrolle der Entwicklung. Bei bestimmten Konstellationen z.B. einer groß-zystischen CPAM, dem Auftreten von Symptomen oder Wachstum der CPAM könnte jedoch eine Operation notwendig werden, um potenzielle Risiken zu minimieren. Entscheidungen über die Behandlung können in enger Zusammenarbeit mit einem unserem spezialisierten Team getroffen werden.
2.
Chirurgische Entfernung: Bei größeren oder symptomatischen CPAMs wird häufig eine chirurgische Entfernung empfohlen. Die Operation, oft eine Lobektomie genannt, beinhaltet die Entfernung des betroffenen Lungenlappens. Dies ist in der Regel die bevorzugte Behandlungsmethode, um potenzielle Komplikationen wie Infektionen oder Atembeschwerden zu vermeiden.
Hier ein Überblick über die chirurgische Therapie einer CPAM
A. Lobektomie: Dies ist die gängigste chirurgische Methode, bei der der betroffene Lungenlappen, der die CPAM enthält, vollständig entfernt wird. Diese Methode wird bevorzugt, weil sie das Risiko von Rezidiven (Wiederkehr der Malformation und damit Gefahr von Infektion und bösartiger Entartung) minimiert und andere Teile der Lunge unberührt lässt.
B. Segmentresektion: Bei dieser Methode wird nur ein Segment des betroffenen Lungenlappens entfernt, nicht der gesamte Lappen. Dies wird manchmal angewandt, wenn die CPAM kleiner ist oder in einem Bereich lokalisiert ist, der eine präzisere Entfernung erlaubt.
C. Thorakoskopie (minimalinvasive Chirurgie): Bei fast allen Kindern kann die Operation in unserer Klinik auch minimalinvasiv durchgeführt werden, was bedeutet, dass nur kleine Schnitte gemacht werden und spezielle Instrumente zusammen mit einer Kamera verwendet werden, um die CPAM zu entfernen. Diese Methode führt in der Regel zu einer schnelleren Erholung und weniger Schmerzen im Vergleich zur offenen Chirurgie.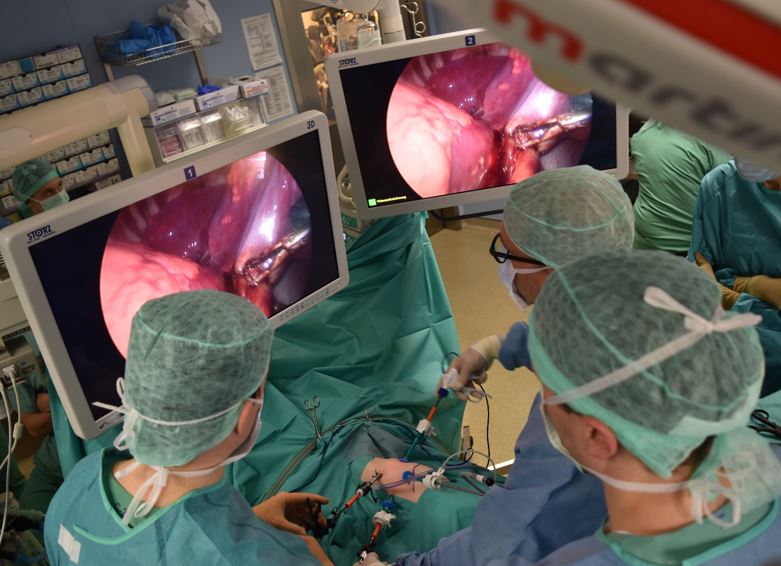
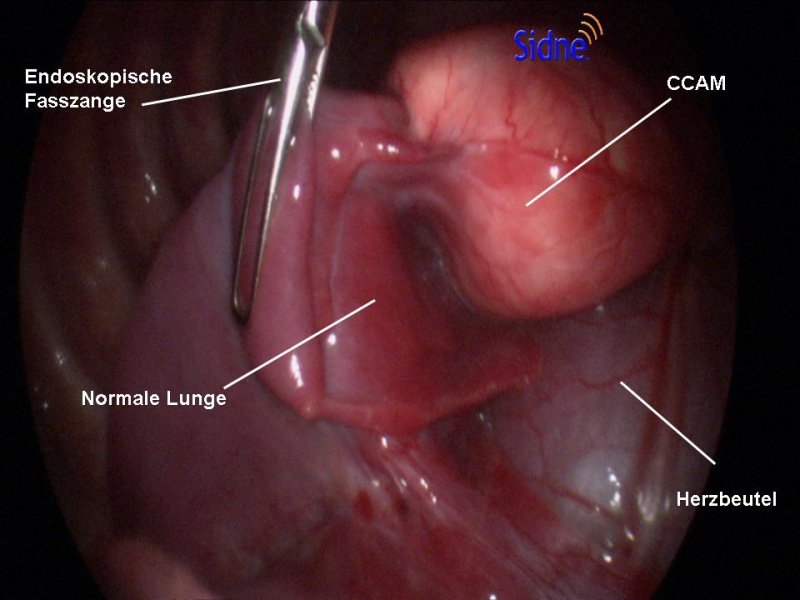
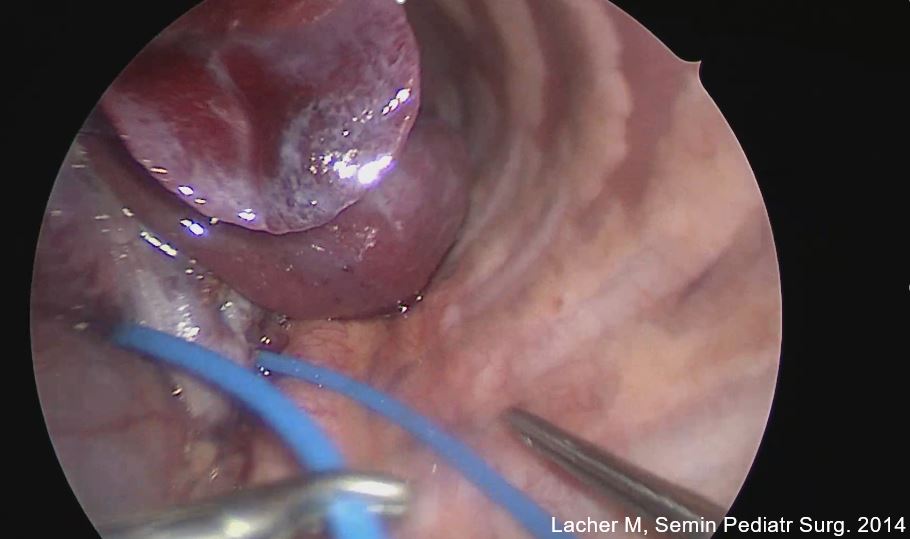
D. Offene Chirurgie: In komplexeren, meist symptomatischen Fällen oder wenn die CPAM sehr groß ist, kann eine offene chirurgische Methode notwendig sein. Dies erfordert größere Schnitte und hat eine längere Erholungszeit, ermöglicht es dem Chirurgen jedoch, die Lunge direkt zu visualisieren und präziser zu arbeiten. In unserem EAT Zentrum kann die Operation auch an der Herz-Lungen-Maschine durchgeführt werden.
3. Pränatale Intervention: In seltenen Fällen, wenn eine CPAM im Ultraschall als besonders groß erscheint und zu einer hydrops fetalis (einer schweren Erkrankung, die durch Flüssigkeitsansammlungen im Fötus gekennzeichnet ist) führt, kann eine pränatale Intervention notwendig sein. Dies könnte eine intrauterine Chirurgie oder andere Maßnahmen umfassen, um die Flüssigkeitsansammlungen zu reduzieren und die Lungenentwicklung zu unterstützen.
4. Postnatale Betreuung: Nach der Geburt benötigen Babys mit CPAM eine sorgfältige Überwachung, um sicherzustellen, dass keine Atemprobleme auftreten. Dies kann die Verwendung von Unterstützungen wie Sauerstoffzufuhr oder sogar mechanische Beatmung in schweren Fällen umfassen.
In jedem Fall empfehlen wir allen Eltern und Betreuern, eng mit unserem Team aus Spezialisten zusammenarbeiten, darunter Geburtsmediziner („Pranatalmedizin)" Neonatologen, Kinderchirurgen und Pädiater, um den besten Behandlungsplan für Ihr Kind zu entwickeln und dessen Gesundheit und Entwicklung zu sichern.